
The Sequence 2/6-2/12
The Human Pangenome, Spectrum of Genes Associated with Childhood-onset Schizophrenia, Evidence of Infection in the 5th to 8th Centuries, New Genetic Variant Associated with Late-onset Ataxia

The human pangenome
The human reference genome is an accepted version of the human genome sequence that is used by researchers as a standard to compare DNA sequences generated in their studies.
Cool. How is it used?
When DNA is sequenced in order to identify mutations, or harmful change in the DNA, the process goes like this: the DNA is fragmented into short segments → these fragments are sequenced —> and the resulting sequence reads are reassembled by aligning to the reads of the standard reference sequence. Potential mutations are then identified by finding variations in the DNA that differ from the reference genome. These differences can be in the form of single nucleotide polymorphisms, or single letter changes, insertions and deletions of letters, or larger structural changes.
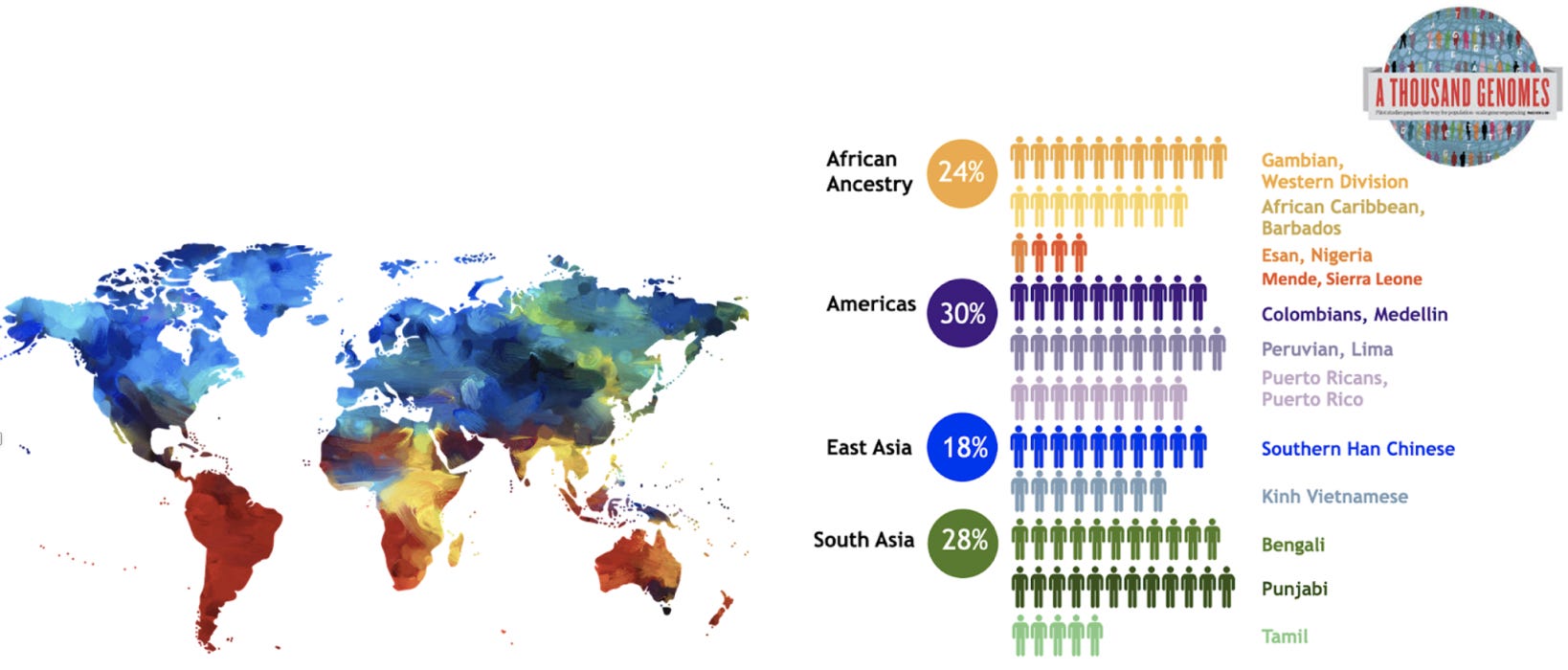
Image credit: The Human Pangenome Reference Center; diagram of the initial sampling effort of 200 samples included in the Pangenome project.
Got it. Where does the ‘pangenome’ come in?
The thing is, the current reference genome is based on the DNA from just a few people from Buffalo, NY. Why? No reason, really, other then they were some of the first successful DNA sequences following the completion of the Human Genome Project, and were of particularly high quality. A problem, because people who share less ancestry with these individuals from Buffalo (i.e. are not white European) benefit less from genetic testing and gene discovery. Current DNA sequencing is biased toward the variation that is in the reference genome; so, when someone of different ancestry is tested, they are likely to have quite a bit of variation that is missing from the reference genome. In fact, in analyzing samples from 154 people around the world, researchers found and found 60Mb (Mb stands for ‘Megabases’, and 1 Mb = 1 million bases) worth of genome content missing from the reference. That is a lot. Additionally, DNA variations appear to show different frequency patterns in different populations. Thinking about the process above, this means that ‘alignment’ stage will be off, missing regions of the genome entirely.
This being said, researchers are considering a new ‘pangenome’ reference genome. A draft was already published here. This draft is made up of 47 genomes, with the goal of ultimately including the genomes of 350 individuals, the vast majority of the genomes being of African ancestry.
What’s the takeaway?
As we move into an era of precision medicine with the ability to treat conditions based on genetic data, it is of the utmost importance to be able to gather accurate genetic testing data from everyone out there. Understanding the diversity of all populations through genetic data will help us to connect genetic variation and rare disease.
Spectrum of genes associated with childhood-onset schizophrenia identified in Jewish population
Alkelai et al. analyzed the DNA of 37 Israeli Jewish families with a proband diagnosed with childhood-onset schizophrenia (COS) in order to understand the genetic spectrum of COS.
What is COS?
Childhood-onset schizophrenia (COS) is a rare form of schizophrenia, a disorder characterized by a range of problems with thinking, behavior or emotions with an onset prior to 13 years of age. Childhood schizophrenia is essentially the same as schizophrenia in adults, but it starts early in life — generally in the teenage years — and has a profound impact on a child's behavior and development. Although COS has been thought to have a ‘multifactorial cause’, i.e. is caused by a combination of genetics as well as the environment, we know that children with a first-degree relative who has the disorder have a five to 20 times higher risk for COS than the general population, solidifying this genetic component.
What do you mean they studied the ‘genetic spectrum’ of COS?
Schizophrenia is believed to be caused by multiple mutations, or harmful changes in the DNA, in different genes and different genomic loci. Understanding the ‘genetic spectrum’ refers to understanding which genes might independently cause COS, and whether they cause more severe or more early onset-disease (aka genotype-phenotype correlation).
So what did they find?
First off, genetic mutations causing schizophrenia were identified in ~20% of the cohort of 37 affected patients (FYI I would consider this a high diagnostic rate for genetic testing in patients with schizophrenia). Also FYI, 14 of those patients had a family history of a first-degree relative with schizophrenia. Makes sense.
The implicated genes identified were ANKRD11, GRIA2, CHD2, CLCN3, CLTC, IGF1R and MICU1. GRIA2 has been established as a potential gene associated with COS previously. Additionally, the team found an increase in rare loss of function (LoF) variants, or variants that cause a loss of functional protein, in the patients with COS.
What’s the takeaway here?
Given these discoveries, it seems genetic testing may have utility in patients with COS. Genetic testing in this population can yield causative genetic diagnoses and provide the ability to assess risk for COS in other family members.
Evidence of infection identified in individuals buried in the 5th to 8th centuries
In a careful extraction of samples from individuals buried in the 5th to 8th centuries at an early medieval Lauchheim settlement in current-day Germany, a team at Kiel University and other centers in Germany and the UK identified bacteria and viruses belonging to infections such as hepatitis B virus (HBV), parvovirus B19 (B19), variola virus (VARV), and Mycobacterium leprae (M. leprae).
How?
The group performed a systematic pathogen screening on the skeletal remains of 70 individuals. Once pathogens for 4 the different viruses (hepatitis B virus, parvovirus B19, variola virus, and Mycobacterium leprae) were identified, the team reconstructed full versions of each viral genome.
Ok got it. What did they find?
Of the 70 individuals sequenced, 22 individuals, or around one-third, were positive for at least one of these infections at their time of death. Seven of the individuals had concurrent infections with two viruses, and one individual had a triple infection of HBV, B19, and M. leprae.
Let’s break down some more of this data:
HBV infection: Identified in 7% of adults and 26.6% of children (please note that ‘children’ here means they weren’t fully-grown adults; in this context, it doesn’t necessarily mean under 18 years old). Today, this prevalence would be considered endemic. The HBV genomes identified were consistent with sub-genotypes that have been previously identified as belonging to 11th to 12th centuries in Germany. Makes sense. However, these days, this sub-genotype is mainly found in Australasia and Northern America, pointing to a change in the geographical distribution of HBV over time.
B19 infections: Identified in 30% of the Lauchheim community. Of the 21 B19-positive individuals, 13 died before reaching an estimated 30 years of age, suggesting premature death could have possibly occurred due to complications developed after a B19 infection. The B19 genomes identified were consistent with the geographic locations of modern and ancient B19 genomes recovered to date.
VARV: Identified in one individual. The VARV genome was also consistent with the known diversity of early medieval VARVs from northern and eastern Europe, and differs from the more modern strains of VARV.
M. leprae: Identified in one adolescent male. Leprosy mainly affects the skin, mucous membranes, and peripheral nerves, however secondary infections caused by Leprosy can lead to deformation and in some cases even loss of extremities. The skeleton of the individual identified exhibited signs of chronic leprosy such as visible facial disfigurement. The researchers concluded that as this adolescent was buried together with the rest of the community, he was likely included in the community and not ostracized, even given his condition. The strain of leprosy identified is the oldest found in Germany to date.
What’s the takeaway?
The infections identified in this settlement indicate a broad distribution of infections across the population at this time. These infections, as well as skeletal lesions identified on the majority of the skeletons, suggest that the community suffered from poor health. The community’s poor health could have been simply caused by the high burden of infectious diseases in the community, compromising the immune system. However, there is a possibly it was due to malnutrition as result of climate change that Europe experienced between the 5th and 7th centuries. Drops in temperature caused crop failure and famine that may have led to this malnutrition. Looking at the big picture, the study suggests that major climate change is a potential driver of overall poor health status of communities.
New genetic variant associated with late-onset ataxia identified
Pellerin et al. studied the DNA of six people from three French Canadian families with autosomal dominant late-onset cerebellar ataxias (LOCAs) in order to identify whether there are unknown changes in the DNA that are linked with it.
Tell me more.
Late-onset cerebellar ataxia (LOCA) is a progressive disorder causing poor coordination of gait, or walking, as well as hands, speech and eye movements. This happens due to degeneration of the cerebellum over time.
There are many types of neurological conditions that cause ataxia, including many sub-types of spinocerebellar ataxia. One of them, spinocerebellar ataxia 27 (SCA27), is an autosomal dominant cerebellar ataxia normally onset in late-childhood to early-adulthood. It is characterized by ataxia with tremor, involuntary movements of the face, psychiatric symptoms and cognitive deficits. Importantly to this story, it associated with variants in the FGF14 gene.
So what did they find?
By analyzing the DNA of these three families and seeing which family members with and without LOCA share and don’t share genetic variants, the team identified a new mutation they believe is the cause of the condition in these families. That variant is a repeat expansion, literally a repeat of the letters ‘GA’, in the FGF14 gene.
You may be saying, ‘I thought we knew FG14 was associated with ataxia’. You would be correct. This is important news for a couple of reasons: Firstly, FG14 was associated with ataxia normally onset in late-childhood to early-adulthood, and not late-onset ataxia. This is what we call a ‘phenotype expansion’ in genetics. Secondly, the mutation identified is not so easy to find. It is in what we call an ‘intronic’ region of the DNA, meaning a part of the DNA that does not code for protein that we know of. In fact, it can only be picked up with whole genome sequencing. Additionally, mutations that are ‘repeats’ like this one, can only really be picked up with what we call ‘long-read sequencing’, or sequencing, which literally reads longer sequences of DNA at a time than traditional DNA sequencing.
What’s the takeaway?
There are surely genetic causes of many conditions out there that have not been discovered. With further studies, significant genetic mutations identified in research like this can be used to help people estimate their risks to pass on conditions like late-onset ataxia. This can lead to either first the appropriate kind of genetic testing needed to identify the mutation, and secondly, opens the door to potential clinical trials that are dependent on a genetic diagnosis. In the case of LOCA, these patients are now eligible for a clinical trial of a drug called aminopyridine, which may reduce symptoms and improve quality of life.